Whole genome assemblies for Triatominae, the vectors of Chagas disease
The process by which insects adapt, to a new food source – blood –, and to a new ecotope – human dwellings -, are some of the most important phenomena in medical entomology. Understanding the genomic basis of the adaptations for hematophagy and domiciliation is of paramount importance for control, to generate targeted interventions against hematophagous vectors.
Trypanosoma cruzi is a protozoan parasite that infects triatomine insect vectors and mammals, causing Chagas disease in humans, a neglected cardiac and digestive chronic illness endemic in the Americas that kills around 10,000 people each year.
Triatomines are hematophagous vectors, grouped in the Subfamily Triatominae, which in turn is part of the Family Reduviidae, which encompasses insects that feed on other insects. Within the triatomines there are species that inhabit sylvatic ecotopes, while others have adapted to peridomiciliary and domiciliary ecotopes.
To study their evolution within Reduviidae and their adaptation, methodologies that provide high resolution by means of including many genetic markers simultaneously are required. The genomic approach and new generation sequencing technologies are powerful tools for this task.

From left to right, Rhodnius ecuadoriensis adults and the five nymphal instars.
Only three whole genome sequences with different degree of completion are currently available, from Rhodnius prolixus, Triatoma rubrofasciata and Triatoma infestans, providing just a glimpse of the genomic particularities of these vectors.
In our project, we aim to fill the whole genome gap in the Triatominae and contribute to the understanding of triatomine biology. We are particularly interested in evaluating the evolution of hematophagy, and their adaptation to the built environment. By comparative genomics, we will analyse the genome of selected triatomine species.
To accomplish this, we have collaborators from several countries of Latin America, providing triatomine samples and genomic information required to generate the assemblies and annotation of several triatomines species.
So far, we are processing the Rhodnius ecuadoriensis and Panstrongylus geniculatus, and once these are finished we will publish them in public repositories and scientific publications.



Trypanosoma cruzi landscape genetics: Ecuador and Peru
Neglected Tropical Diseases affect approximately 2.7 billion people worldwide and are widely distributed, especially among the tropics. The dispersal and distribution of the pathogens that cause NTDs are strongly influenced by the spatial complexity of their physical and biotic environment. Thus, spatial analyses of abiotic and biotic variables can serve to accurately predict the emergence, occurrence, and spread of disease.
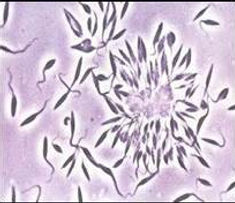

Trypanosoma cruzi (left) and the local vector species Rhodnius ecuadoriensis (right).
In this project we focus on Chagas disease in an area of disease transmission in Southern Ecuador and Northern Peru. Chagas disease is caused by the single-celled kinetoplastid Trypanosoma cruzi, and in our study region the local insect vector species is Rhodnius ecuadoriensis. Our aim is to establish the population genetic/genomic diversity of both vector and parasite across the study region by high resolution (genome scale) genotyping. To evaluate the interconnectivity of different vector and parasite populations, and to predict potential dispersal routes or barriers to geneflow, we adopt a landscape genetics approach.
Our aim is to define what environmental factors and what spatial scales are relevant in defining parasite and vector population distribution. Such descriptions of diversity are key to measuring and predicting dispersal. Our evaluation of barriers to gene flow will feed directly into a rationale for effective local disease control.
